
News
>
News >
News>
3D Medicines Announced Clinical Trial Data for Subcutaneous PD-L1 Antibody KN035 on CSCO 2019 Annual Meeting
3D Medicines Announced Clinical Trial Data for Subcutaneous PD-L1 Antibody KN035 on CSCO 2019 Annual Meeting
2019-09-19 00:00:00
Source:思路迪
Author:思路迪医药
Views:1507
On September 19, 2019, 3D Medicines Inc. attended the 22nd annual meeting of Chinese Society of Clinical Oncology (CSCO). Toshio Shimizu, M.D., PhD. from National Cancer Center Hospital, gave an oral presentation on the Clinical Trial Periodic Report of the phase I study for Envafolimab (KN035), a subcutaneous anti-PD-L1 single domain antibody from 3D Medicines, in advanced solid tumor patients in Japan. Details of the presentation are listed as follows:
Oral Presentation
Title: Phase I Study of Envafolimab (KN035), the First Subcutaneously Administered, Novel Fusion Anti-PD-L1 Antibody in Japanese Patients with Advanced Solid Tumors
Speaker: Toshio Shimizu, M.D., PhD.
Time: September 19, 2019, 16:25-16:35
Venue: Session 3 on Safety Management for Anti-tumor Drugs, 3rd State Banquet Hall, 2nd Floor, Xiamen International Conference Center

The detailed information of the published results is as follows:
Background
KN035 is a novel fusion protein of humanized anti-PD-L1 single domain antibody and human IgG1 Fc independently invented in China, which has significant differentiation advantages over PD1/PD-L1 antibodies in development or on the market. The advantages of Envafolimab (KN035) include administration by subcutaneous (SC) injection, stability at room temperature and rapid tumor penetration, which improves patient compliance and their quality of life, as well as conforming to the trend of managing cancer as chronic illness. Up to now, the clinical trials for Envafolimab (KN035) have been proceeding smoothly in the United States, Japan and China, and it has demonstrated preliminary efficacy with a favorable safety profile in solid tumor patients who have failed multi-line therapies.
The results published on the CSCO annual meeting come from an open-ended dose-escalation and dose-expansion phase I clinical trial in advanced solid tumor patients in Japan, which aims to evaluate the safety, tolerability, pharmacokinetics (PK) and preliminary anti-tumor efficacy of Envafolimab (KN035).
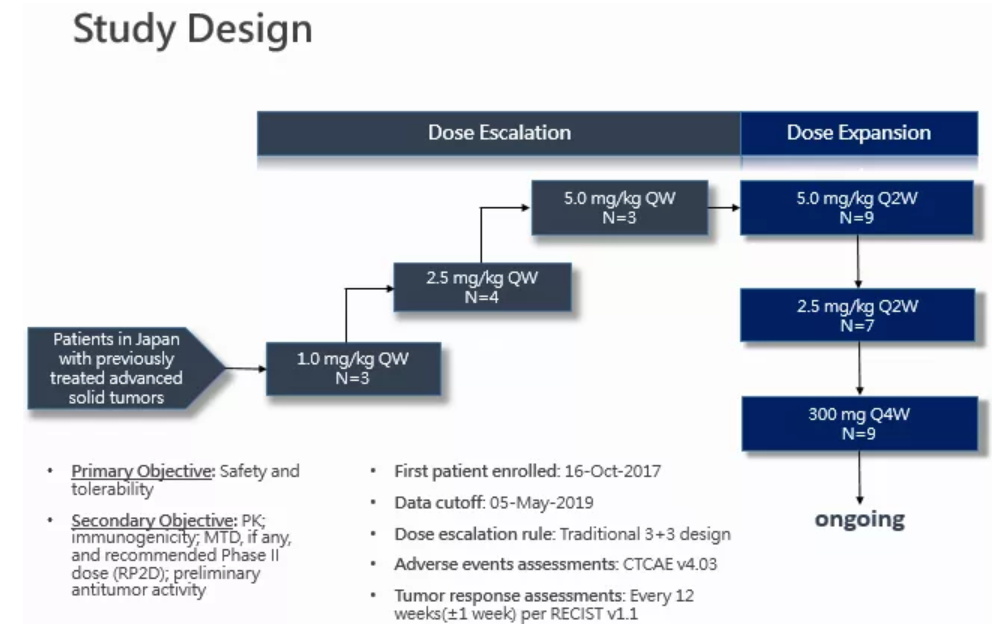
Study Design and Data Analysis
The study is a dose-escalation and dose-expansion phase I clinical trial to evaluate the safety, tolerability, pharmacokinetic characteristics and preliminary anti-tumor efficacy of Envafolimab (KN035) monotherapy in advanced solid tumor patients in Japan. Key inclusion criteria include advanced solid tumors confirmed by histology or cytology examinations, failure or intolerance of standard treatment, unsuitability for surgery or local treatment, ECOG 0-1, and adequate hematopoietic and organ function.
The dose-escalation phase followed a traditional 3+3 design and the dose levels were planned at 1.0, 2.5, 5.0 mg/kg SC once every-7-days (QW) with dose limiting toxicity (DLT) evaluation on the first cycle of treatment period (within 28 days of first dosing). The dose-escalation would continue till maximum tolerated dose (MTD) was seen, and the recommended dose for following clinical trials would be thereby decided. As for dose-expansion, patients were treated with Envafolimab (KN035) SC at 2.5, 5.0 mg/kg once every-14-days (Q2W), or a fixed dose (300mg) once every-4-weeks (Q4W), for the purpose of further study of the safety, pharmacokinetics and preliminary efficacy in patients with locally advanced or metastatic solid tumors. Patients of the fixed-dose group are still being enrolled, so no results have been obtained.
The first patient was enrolled on October 16, 2017. As of May 5, 2019, 26 advanced solid tumor patients (dose-escalation group (n=10), dose-expansion group (n=16)) have been enrolled, including patients with biliary tract cancer (n=4), soft tissue sarcoma (n=3), urothelial carcinoma (n=2), ovarian cancer (n=2), colorectal cancer (n=2), pancreatic cancer (n=2) and other solid tumors (n=11). The median age of the patients was 59 years, 42.3% of them were males, and 88.5% had received second or later lines of treatment. As of the data cut-off date, 23 patients have withdrawn from the trial (21 patients withdrew due to disease progression), and 3 patients were still receiving treatment. Response was assessed per RECIST 1.1 every 12 weeks and 12 patients had received post-baseline tumor efficacy evaluation at least once.
Results
Safety
No DLT was observed up to the highest dose level of 5 mg/kg QW. No maximum tolerated dose (MTD) was reached.
Regardless of severity and relevance to treatment, the most frequent adverse events (AEs) included fever (19.2%), low lymphocyte count (15.4%), itching (15.4%), rash (15.4%), headache (15.4%) and vomiting (11.5%). The most frequent drug-related treatment emergent adverse events (TEAEs) included fever (11.5%), itching (11.5%) and rash (11.5%), all of which were grade 1 or 2. No grade 5 drug-related TEAEs were reported. 1 patient in the 5.0 mg/kg Q2W group experienced a grade 3 drug-related serious adverse event (cerebral infarction, which may be related), and 1 patient in the 2.5 mg/kg Q2W group experienced a grade 2 drug-related serious adverse event (rash, resolved), which was classified by the investigator as an immune-mediated adverse event.
Judging from the safety data from the phase I trial in Japan, Envafolimab (KN035) has demonstrated an overall manageable safety profile.
Preliminary Efficacy
As of May 5, 2019, 12 patients had been evaluated for efficacy as shown in the figure below, (11 are shown in the waterfall plot, and 1 had no target lesion at baseline), 4 patients were observed with response up to partial response (PR), and 3 patients (25.0%) were confirmed. Patients with PR included 1 patient with esophageal cancer and 1 with ovarian cancer, both at 5.0mg/kg Q2W. The time to progression (TTP) in the esophageal cancer patient was 44.4 weeks, and the duration of response (DOR) was 32.7 weeks. The time to response (TTR) in the ovarian cancer patient was 24.6 weeks, and the patient withdrew from the trial due to adverse events. 1 urothelial carcinoma patient was from the 1.0 mg/kg QW group, the TTP was 53.1 weeks and the DOR was 41.9 weeks; 1 extrahepatic bile duct cancer patient (efficacy not confirmed) was from the 5.0 mg/kg QW group, the TTP was 16.3 weeks and the DOR was 5.1 weeks. Except for the above-mentioned patient with ovarian cancer, all the patients with PR showed significant tumor regression at the first efficacy evaluation 12 weeks after the administration.
Response up to stable disease (SD) was observed in 5 patients (41.7%), including 1 urothelial carcinoma patient from the 1.0 mg/kg QW group (no target lesion at baseline, best efficacy evaluated by the investigator was SD), who had received 19 cycles of treatment, and was still receiving treatment as of the data cut-off date; 1 appendix cancer patient from the 2.5 mg/kg QW group; 1 ovarian cancer patient from the 2.5 mg/kg Q2W group; 1 neuroendocrine tumor patient and 1 carcinoma with unknown primary (CUP) patient, both from the 5.0 mg/kg Q2W group. The median duration of SD among the 4 patients mentioned above was 11.5 weeks.
Most of the subjects have received second or later lines of treatment, who were left with few treatment options and poor prognosis. For these advanced solid tumor patients, who were not selected with a biomarker-driven approach, Envafolimab (KN035) monotherapy demonstrated favorable efficacy, with the objective response rate (ORR) in the efficacy-evaluable population at 25.0% and the disease control rate (DCR) at 66.7%. Lasting high response to the treatment was observed in some of the patients, showing the possibility that Envafolimab (KN035) has provided a brand-new promising treatment option for patients.

Pharmacokinetic Characteristics
As is shown in the graph below, preliminary PK analysis has demonstrated excellent stability and long half-life of Envafolimab (KN035), which supports lower administration frequency in future clinical trials. Exposures of KN035 increased approximately proportionally with dose in both QW and Q2W schedules.
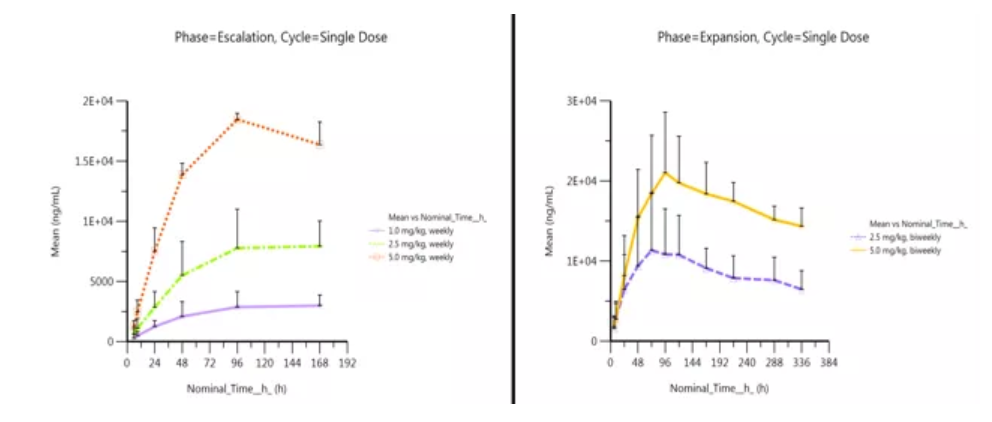
Left: The plasma levels-nominal time graph of patients in the dose-escalation group after QW single-dose
Right: The plasma levels-nominal time graph of patients in the dose-expansion group after Q2W single-dose
Conclusions
Envafolimab (KN035) monotherapy exhibits good tolerability with a favorable safety profile in patients with multiple advanced solid tumors in Japan. No DLT was observed up to the highest dose level and MTD was not reached, demonstrating encouraging anti-tumor activity and duration of response. Based on PK data from the Q2W schedule, the fixed dose of the Q4W schedule is presently being evaluated.
Overall, Envafolimab (KN035), the world’s first subcutaneous anti-PD-L1 single domain antibody, has demonstrated a safety profile and preliminary efficacy comparable to similar intravenous products, with significant efficacy, prolonged response and convenient clinical use. In consideration of the favorable phase I data, Envafolimab (KN035) is currently going through other clinical trials in China - a phase II trial in MSI-H solid tumors and a phase III trial in biliary tract cancer.
热门文章




如您想了解更多
请咨询我们
请咨询我们
